
 请用手机扫二维码
请用手机扫二维码
医薬品情報
| 総称名 | スマイラフ |
|---|---|
| 一般名 | ペフィシチニブ臭化水素酸塩 |
| 欧文一般名 | Peficitinib Hydrobromide |
| 製剤名 | ペフィシチニブ臭化水素酸塩錠 |
| 薬効分類名 | ヤヌスキナーゼ(JAK)阻害剤 |
| 薬効分類番号 | 3999 |
| ATCコード | L04AA49 |
| KEGG DRUG | D10721 ペフィシチニブ臭化水素酸塩 |
| KEGG DGROUP | DG02020 JAK阻害薬 DG01985 疾患修飾性抗リウマチ薬 (DMARD) |
| JAPIC | 添付文書(PDF) |
添付文書情報2021年10月 改訂(第2版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| スマイラフ錠50mg | Smyraf Tablets 50mg | アステラス製薬 | 3999046F1023 | 1547.4円/錠 | 劇薬, 処方箋医薬品 |
| スマイラフ錠100mg | Smyraf Tablets 100mg | アステラス製薬 | 3999046F2020 | 3030.4円/錠 | 劇薬, 処方箋医薬品 |
1. 警告
1.1 本剤投与により、肺炎、敗血症、ウイルス感染等による重篤な感染症の新たな発現若しくは悪化等が報告され、本剤との関連性は明らかではないが、悪性腫瘍の発現も報告されている。本剤が疾病を完治させる薬剤でないことも含め、これらの情報を患者に十分説明し、患者が理解したことを確認した上で、治療上の有益性が危険性を上回ると判断される場合にのみ投与すること。また、本剤投与により重篤な副作用が発現し、致死的な経過をたどることがあるので、緊急時の対応が十分可能な医療施設及び医師が使用し、本剤投与後に副作用が発現した場合には、主治医に連絡するよう患者に注意を与えること。[1.2.1、1.2.2、2.1、2.2、7.2、8.1-8.5、8.9、9.1.1-9.1.3、9.1.10、11.1.1、15.1.1-15.1.3参照]
1.2 感染症
1.2.1 重篤な感染症
敗血症、肺炎、真菌感染症を含む日和見感染症等の致死的な感染症が報告されているため、十分な観察を行うなど感染症の発症に注意すること。[1.1、2.1、7.2、8.1、8.9、9.1.1、9.1.3、11.1.1、15.1.1参照]
1.2.2 結核
播種性結核(粟粒結核)及び肺外結核(脊椎、脳髄膜、胸膜、リンパ節等)を含む結核があらわれる可能性がある。結核の既感染者では症状の顕在化及び悪化のおそれがあるため、本剤投与に先立って結核に関する十分な問診及び胸部レントゲン検査に加え、インターフェロン-γ遊離試験又はツベルクリン反応検査を行い、適宜胸部CT検査等を行うことにより、結核感染の有無を確認すること。結核の既往歴を有する患者及び結核の感染が疑われる患者には、結核等の感染症について診療経験を有する医師と連携の下、原則として本剤の投与開始前に適切な抗結核薬を投与すること。また、ツベルクリン反応等の検査が陰性の患者において、投与後活動性結核があらわれる可能性がある。[1.1、2.2、8.2、9.1.2、11.1.1参照]
1.3 本剤の治療を行う前に、少なくとも1剤の抗リウマチ薬等の使用を十分勘案すること。また、本剤についての十分な知識とリウマチ治療の経験をもつ医師が使用すること。
2. 禁忌
次の患者には投与しないこと
2.7 本剤の成分に対し過敏症の既往歴のある患者
2.8 妊婦又は妊娠している可能性のある女性[9.5参照]
4. 効能または効果
既存治療で効果不十分な関節リウマチ(関節の構造的損傷の防止を含む)
5. 効能または効果に関連する注意
過去の治療において、メトトレキサートをはじめとする少なくとも1剤の抗リウマチ薬等による適切な治療を行っても、疾患に起因する明らかな症状が残る場合に投与すること。
6. 用法及び用量
通常、成人にはペフィシチニブとして150mgを1日1回食後に経口投与する。なお、患者の状態に応じて100mgを1日1回投与できる。
7. 用法及び用量に関連する注意
8. 重要な基本的注意
8.1 本剤は、免疫反応に関与するJAKファミリー(JAK1/JAK2/JAK3/TYK2)を阻害するので、感染症に対する宿主免疫能に影響を及ぼすおそれがある。本剤の投与に際しては十分な観察を行い、感染症の発現や増悪に注意すること。また、患者に対し、発熱、倦怠感等があらわれた場合には、速やかに主治医に相談するよう指導すること。[1.1、1.2.1、2.1、7.2、8.9、9.1.1、9.1.3、11.1.1、15.1.1、15.2.1参照]
8.2 本剤投与に先立って結核に関する十分な問診及び胸部レントゲン検査に加え、インターフェロン-γ遊離試験又はツベルクリン反応検査を行い、適宜胸部CT検査等を行うことにより、結核感染の有無を確認すること。また、本剤投与中も胸部レントゲン検査等の適切な検査を定期的に行うなど結核の発現には十分に注意し、患者に対し、結核を疑う症状が発現した場合(持続する咳、発熱等)には速やかに主治医に連絡するよう説明すること。[1.1、1.2.2、2.2、9.1.2、11.1.1参照]
8.4 ヘルペスウイルスを含むウイルスの再活性化(帯状疱疹等)が報告されている。また、重篤な帯状疱疹や播種性帯状疱疹も認められていることから、ヘルペスウイルス等の再活性化の徴候や症状の発現に注意すること。徴候や症状の発現が認められた場合には、患者に受診するよう説明し、本剤の投与を中断し、速やかに適切な処置を行うこと。また、ヘルペスウイルス以外のウイルスの再活性化にも注意すること。[1.1、11.1.1、15.2.1参照]
8.6 好中球減少、リンパ球減少及びヘモグロビン減少があらわれることがあるので、本剤投与開始後は定期的に好中球数、リンパ球数及びヘモグロビン値を確認すること。[2.4-2.6、9.1.5-9.1.7、11.1.2、15.2.1参照]
8.7 総コレステロール、LDLコレステロール、HDLコレステロール及びトリグリセリドの上昇等の脂質検査値異常があらわれることがある。本剤投与開始後は定期的に脂質検査値を確認すること。臨床上必要と認められた場合には、脂質異常症治療薬の投与等の適切な処置を考慮すること。
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
9.1.2 結核の既感染者(特に結核の既往歴のある患者及び胸部レントゲン上結核治癒所見のある患者)及び結核感染が疑われる患者
9.1.3 易感染性の状態にある患者
9.1.4 腸管憩室のある患者
消化管穿孔があらわれるおそれがある。[11.1.3参照]
9.1.5 好中球減少のある患者
9.1.6 リンパ球減少のある患者
9.1.7 ヘモグロビン減少のある患者
9.1.8 間質性肺炎の既往歴のある患者
定期的に問診を行うなど、注意すること。間質性肺炎があらわれるおそれがある。[11.1.5参照]
9.1.9 先天性QT短縮症候群の患者
治療上の有益性が危険性を上回ると判断される場合にのみ投与すること。QT間隔が短縮するおそれがある。[17.3.1参照]
9.1.10 B型肝炎ウイルスキャリアの患者又は既往感染者(HBs抗原陰性、かつHBc抗体又はHBs抗体陽性)
9.3 肝機能障害患者
9.3.1 重度の肝機能障害を有する患者
9.3.2 中等度の肝機能障害を有する患者(Child-Pugh分類B)
9.3.3 軽度の肝機能障害を有する患者(Child-Pugh分類A)
9.4 生殖能を有する者
妊娠可能な女性に投与する場合には、投与中及び投与終了後少なくとも1月経周期は適切な避妊を行うよう指導すること。[9.5参照]
9.5 妊婦
妊婦又は妊娠している可能性のある女性には投与しないこと。動物実験ではラットで催奇形性、ウサギで胚・胎児致死作用が報告されており、ヒトに本剤を投与したときの血漿中濃度と比較したとき、胚・胎児発生に関する安全域はラット及びウサギでそれぞれ1.2倍及び0.9倍であった。また、ラットで胎児の発達への影響、出生児の生存率、体重への影響及び骨格奇形が報告されている。雌ラットの受胎能及び初期胚発生に関する安全域は3.6倍、出生前及び出生後の発生に関する安全域は0.7倍であった1)。[2.8、9.4参照]
9.6 授乳婦
9.7 小児等
小児等を対象とした有効性及び安全性を指標とした臨床試験は実施していない。
9.8 高齢者
用量に留意して、患者の状態を観察しながら慎重に投与すること。一般に、生理機能が低下している。また、重篤な感染症の発現率の上昇が認められている。
10. 相互作用
10.2 併用注意
11. 副作用
11.1 重大な副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1.1 感染症
帯状疱疹(12.9%)、肺炎(ニューモシスチス肺炎等を含む)(4.7%)、敗血症(0.2%)等の重篤な感染症があらわれることがある。本剤投与中に重篤な感染症を発現した場合は、感染症がコントロールできるようになるまでは投与を中止すること。[1.1、1.2.1、1.2.2、2.1、2.2、7.2、8.1-8.4、8.9、9.1.1-9.1.3、9.1.10、15.1.1参照]
11.1.2 好中球減少症(0.5%)、リンパ球減少症(5.9%)、ヘモグロビン減少(2.7%)
本剤投与開始前及び投与中は、定期的に血液検査を行うこと。
好中球数
本剤投与開始後、好中球数が継続して500〜1000/mm3である場合は、好中球数が1000/mm3を超えるまで本剤の投与を中断すること。
リンパ球数
本剤投与開始後、リンパ球数が500/mm3未満になった場合には、500/mm3以上となるまで本剤の投与を中止すること。
ヘモグロビン値
本剤投与開始後、ヘモグロビン値が8g/dL未満になった場合には、正常化するまで本剤の投与を中止すること。
11.1.3 消化管穿孔(0.3%)
異常が認められた場合には投与を中止するとともに、腹部レントゲン、CT等の検査を実施するなど十分に観察し、適切な処置を行うこと。[9.1.4参照]
11.1.4 肝機能障害、黄疸
11.1.5 間質性肺炎(0.3%)
発熱、咳嗽、呼吸困難等の呼吸器症状に十分に注意し、異常が認められた場合には、速やかに胸部レントゲン検査、胸部CT検査及び血液ガス検査等を実施し、本剤の投与を中止するとともにニューモシスチス肺炎との鑑別診断(β-Dグルカンの測定等)を考慮に入れ適切な処置を行うこと。[9.1.8参照]
11.2 その他の副作用
| 5%以上 | 1〜5%未満 | 1%未満 | |
| 感染症及び寄生虫症 | 咽頭炎、上咽頭炎、上気道感染、気管支炎、インフルエンザ、膀胱炎 | 扁桃炎、副鼻腔炎、胃腸炎、結膜炎、中耳炎、足部白癬、歯周炎、歯肉炎、口腔ヘルペス、単純ヘルペス、尿路感染 | |
| 神経系障害 | 頭痛 | ||
| 血管障害 | 高血圧 | ||
| 呼吸器、胸郭及び縦隔障害 | 上気道の炎症、咳嗽、口腔咽頭痛、喘息 | ||
| 胃腸障害 | 悪心、嘔吐、口内炎、齲歯、下痢、便秘、胃炎、胃食道逆流性疾患、上腹部痛、腹部不快感 | ||
| 皮膚及び皮下組織障害 | 湿疹、発疹 | ||
| 筋骨格系及び結合組織障害 | 筋痙縮、背部痛 | ||
| 一般・全身障害及び投与部位の状態 | 発熱、倦怠感 | ||
| 臨床検査 | 血中CK増加、脂質増加 | 白血球数減少、肝機能検査値上昇、血中β-Dグルカン増加、血中コレステロール増加 | AST増加、ALT増加、γ-GTP増加、B型肝炎DNA増加 |
14. 適用上の注意
14.1 薬剤交付時の注意
PTP包装の薬剤はPTPシートから取り出して服用するよう指導すること。PTPシートの誤飲により、硬い鋭角部が食道粘膜へ刺入し、更には穿孔をおこして縦隔洞炎等の重篤な合併症を併発することがある。
15. その他の注意
15.1 臨床使用に基づく情報
15.1.1 臨床試験における重篤な感染症の発現率
第3相試験2試験の併合解析において、報告された100人・年あたりの重篤な感染症の発現率(95%信頼区間)は、本剤100mgで2.8(1.4,5.4)、150mgで3.0(1.6,5.6)、本剤合計で2.9(1.9,4.6)であった。また、後期第2相試験、第3相試験2試験及び継続投与試験の4試験の安全性併合解析において、100人・年あたりの重篤な感染症の発現率(95%信頼区間)は、本剤合計で2.5(1.9,3.2)であった。[1.1、1.2.1、2.1、8.1、11.1.1参照]
15.1.2 臨床試験における悪性腫瘍(非黒色腫皮膚癌を除く)の発現率
第3相試験2試験の併合解析において、報告された100人・年あたりの悪性腫瘍(非黒色腫皮膚癌を除く)の発現率(95%信頼区間)は、本剤100mgで1.2(0.5,3.3)、150mgで0.0、本剤合計で0.6(0.2,1.6)であった。
後期第2相試験、第3相試験2試験及び継続投与試験の4試験の安全性併合解析において、報告された100人・年あたりの悪性腫瘍(非黒色腫皮膚癌を除く)の発現率(95%信頼区間)は、本剤合計で0.9(0.6,1.3)であった。
また、投与期間別の発現状況は下記のとおりであった。[1.1、8.5参照]
後期第2相試験、第3相試験2試験及び継続投与試験の4試験の安全性併合解析において、報告された100人・年あたりの悪性腫瘍(非黒色腫皮膚癌を除く)の発現率(95%信頼区間)は、本剤合計で0.9(0.6,1.3)であった。
また、投与期間別の発現状況は下記のとおりであった。[1.1、8.5参照]
| 投与期間 | 評価例数、曝露期間 | %(例数) | 発現率(/100人・年)(95%信頼区間) |
| 全体 | 1052例、2332.8人・年 | 1.9%(20) | 0.9(0.6,1.3) |
| 0〜6カ月 | 1052例、494.6人・年 | 0.4%(4) | 0.8(0.3,2.2) |
| 6〜12カ月 | 918例、437.3人・年 | 0.4%(4) | 0.9(0.3,2.4) |
| 12〜18カ月 | 826例、388.2人・年 | 0.5%(4) | 1.0(0.4,2.7) |
| 18〜24カ月 | 724例、319.4人・年 | 0.6%(4) | 1.3(0.5,3.3) |
| 24〜36カ月 | 555例、384.3人・年 | 0.4%(2) | 0.5(0.1,2.1) |
| 36〜48カ月 | 237例、149.0人・年 | 0.0%(0) | 0.0 |
| 48〜60カ月 | 110例、101.7人・年 | 1.8%(2) | 2.0(0.5,7.9) |
| 60カ月〜 | 90例、58.4人・年 | 0.0%(0) | 0.0 |
15.1.3 心血管系事象のリスク因子を有する関節リウマチ患者を対象としたJAK阻害剤トファシチニブクエン酸塩の海外臨床試験の結果、主要評価項目である主要な心血管系事象(Major Adverse Cardiovascular Events:MACE)及び悪性腫瘍(非黒色腫皮膚癌を除く)の発現率について、TNF阻害剤群に対するハザード比(95%信頼区間)はそれぞれ1.33(0.91,1.94)及び1.48(1.04,2.09)であり、95%信頼区間上限は予め設定していた非劣性マージン1.8を超え、TNF阻害剤群に対する非劣性が検証されなかったことが報告されている。また、本剤でも、国内市販後の自発報告において、心血管系事象の発現が認められている。[1.1、8.5参照]
15.2 非臨床試験に基づく情報
16. 薬物動態
16.1 血中濃度
16.1.1 単回投与
日本人健康成人(各群6例)にペフィシチニブ20、60、200mgを空腹時単回経口投与したとき注)、ペフィシチニブの血漿中濃度は投与後1〜2時間でピークに達し、消失半減期は3.7〜7.5時間であった5)。
| 投与量 | 被験者数 | Cmax(ng/mL) | Tmax(h) | t1/2(h) | AUCinf(ng・h/mL) |
| 20mg | 6 | 76.87±24.19 | 1.4±0.8 | 3.7±0.7 | 259.50±42.91 |
| 60mg | 6 | 241.13±74.74 | 1.3±0.3 | 4.0±1.0 | 782.78±158.56 |
| 200mg | 6 | 648.73±55.48 | 1.8±0.4 | 7.5±4.9 | 2524.88±234.45 |
16.1.2 反復投与
日本人健康成人(24例)にペフィシチニブ150mgを1日1回食後反復経口投与したとき、反復投与3日目には定常状態に達し、定常状態でのCmaxは613.2ng/mL、AUC24hは2643ng・h/mLであった。また、単回投与時と比較した定常状態での蓄積比は1.2であった6)。
16.2 吸収
日本人健康成人(18例)にペフィシチニブ150mgを単回経口投与したとき、空腹時投与に比べ食後投与ではCmaxは56.4%、AUClastは36.8%増加した7)。
16.3 分布
ペフィシチニブの血漿蛋白結合率は72.83%〜75.20%であり、主要結合蛋白質はアルブミンであった8)(in vitro試験)。
16.4 代謝
ペフィシチニブは主に硫酸抱合代謝を受け、一部はメチル化代謝を受けた9)。ペフィシチニブの主代謝酵素は硫酸転移酵素であるSULT2A1であり、メチル転移酵素であるNNMTも寄与することが示された10)(in vitro試験)。
16.5 排泄
日本人健康成人(各群6例)にペフィシチニブ20、60、200mgを単回経口投与したとき注)、ペフィシチニブの尿中排泄率は12.5%〜16.8%であった5)。
健康成人(6例)に14Cで標識したペフィシチニブ100mgを単回経口投与したとき、放射能として投与量の36.8%が尿中に、56.6%が糞中に排泄された11)(外国人データ)。
16.6 特定の背景を有する患者
16.6.1 腎機能障害患者
軽度(8例)、中等度(8例)、重度(7例)の腎機能障害患者及び腎機能正常被験者(8例)に、ペフィシチニブ150mgを単回経口投与したとき、軽度腎機能障害患者では腎機能正常被験者に比べCmaxは10.4%、AUCinfは12.7%低かった。中等度腎機能障害患者では腎機能正常被験者に比べCmaxは21.7%、AUCinfは16.9%低かった。重度腎機能障害患者では腎機能正常被験者に比べCmaxは21.7%低く、AUCinfは8.7%高かった12)。
| 腎機能障害の程度 | 幾何平均比(90%信頼区間) 腎機能障害患者/腎機能正常被験者 | |
| Cmax | AUCinf | |
| 軽度腎機能障害患者 (60mL/min/1.73m2=<eGFR<90mL/min/1.73m2) | 0.896 (0.595,1.349) | 0.873 (0.610,1.250) |
| 中等度腎機能障害患者 (30mL/min/1.73m2=<eGFR<60mL/min/1.73m2) | 0.783 (0.520,1.179) | 0.831 (0.581,1.190) |
| 重度腎機能障害患者 (15mL/min/1.73m2=<eGFR<30mL/min/1.73m2) | 0.783 (0.513,1.197) | 1.087 (0.738,1.602) |
16.6.2 肝機能障害患者
軽度(8例)、中等度(8例)の肝機能障害患者及び肝機能正常被験者(8例)にペフィシチニブ150mgを単回経口投与したとき、軽度肝機能障害患者では肝機能正常被験者に比べCmaxは3.9%、AUCinfは18.5%高かった。中等度肝機能障害患者では肝機能正常被験者に比べCmaxは82.4%、AUCinfは92.3%高かった13)。[2.3、7.1、9.3.1-9.3.3、10.2、11.1.4参照]
| 肝機能障害の程度 | 幾何平均比(90%信頼区間) 肝機能障害患者/肝機能正常被験者 | |
| Cmax | AUCinf | |
| 軽度肝機能障害患者 (Child-Pugh分類A、スコア5〜6) | 1.039 (0.705,1.531) | 1.185 (0.857,1.638) |
| 中等度肝機能障害患者 (Child-Pugh分類B、スコア7〜9) | 1.824 (1.238,2.686) | 1.923 (1.391,2.658) |
16.7 薬物相互作用
16.7.1 その他の薬剤
(1)ペフィシチニブの薬物動態に及ぼす併用薬の影響
(2)併用薬の薬物動態に及ぼすペフィシチニブの影響
・In vitro試験
ペフィシチニブはCYP3A及びCYP2C8を阻害する17)。また、ペフィシチニブは排出トランスポーターであるBCRP及び取り込みトランスポーターであるOATP1B1及びOCT1を阻害する18)19)20)。
・臨床薬物相互作用試験
| 併用薬 | 併用薬投与量 | ペフィシチニブ投与量 | 幾何平均比(90%信頼区間) 併用/単独 | |
| Cmax | AUC | |||
| ミダゾラムa)(CYP3A基質) | 3mg 単回 | 100mg 1日2回注) | 1.1332 (1.0595,1.2121) | 1.3698 (1.2837,1.4616) |
| ロスバスタチンa)(OATP1B1基質) | 10mg 単回 | 150mg 1日1回 | 1.1484 (1.00741,1.30922) | 1.1826 (1.00386,1.39313) |
| メトホルミン(OCT1、MATE1基質) | 750mg 単回 | 150mg 1日1回 | 0.830 (0.786,0.876) | 0.826 (0.784,0.870) |
| メトトレキサートa) | 15〜25mg 週1回 | 100mg 1日2回注) | 0.9226 (0.8301,1.0254) | 1.0251 (0.9287,1.1315) |
| ミコフェノール酸モフェチルa)、b) | 1000mg 単回 | 100mg 1日2回注) | 0.9457 (0.8003,1.1175) | 1.0248 (0.9619,1.0917) |
| タクロリムスa)(CYP3A基質) | 5mg 単回 | 100mg 1日2回注) | 1.5654 (1.4038,1.7457) | 1.6322 (1.5008,1.7751) |
注)本剤の承認された用法及び用量は「通常、成人にはペフィシチニブとして150mgを1日1回食後に経口投与する。なお、患者の状態に応じて100mgを1日1回投与できる。」である。
17. 臨床成績
17.1 有効性及び安全性に関する試験
17.1.1 国際共同第3相試験(国際共同、単剤若しくはDMARD併用、CL-RAJ3試験)
メトトレキサート(MTX)を含む従来型疾患修飾性抗リウマチ薬(cDMARDs)に対して効果不十分な関節リウマチ患者(目標例数500例(本剤及びプラセボ群各群100例、参照群200例))を対象としたプラセボ対照無作為化二重盲検並行群間比較試験を日本、韓国及び台湾で実施した。cDMARDs併用下若しくは単剤投与下で、本剤100mg、150mg又はプラセボを1日1回朝食後に経口投与した。また参照群として、エタネルセプト50mgを非盲検下、1週間隔で皮下投与した。本剤100mg及び150mg群の投与12週後のACR20%改善率(主要評価項目)はプラセボ群に比べて高く、統計学的に有意な差が認められた25)。[7.1、9.3.2、9.3.3参照]
| 100mg群 | 150mg群 | プラセボ群 | エタネルセプト群(参照群) | |
| 全体集団 | ||||
| ACR20%改善率 | 57.7(60/104) | 74.5(76/102) | 30.7(31/101) | 83.5(167/200) |
| プラセボ群との差[95%信頼区間] | 27.0[12.9,41.1] | 43.8[30.5,57.1] | − | 52.8[41.7,63.9] |
| オッズ比[95%信頼区間]a) p値a)、b) | 3.13[1.76,5.58] <0.001 | 6.59[3.56,12.20] <0.001 | − | |
| ACR50%改善率 | 30.8(32/104) | 42.2(43/102) | 8.9(9/101) | 52.5(105/200) |
| ACR70%改善率 | 13.5(14/104) | 27.5(28/102) | 1.0(1/101) | 30.5(61/200) |
| 日本人部分集団 | ||||
| ACR20%改善率 | 61.2(52/85) | 74.7(62/83) | 28.9(24/83) | 84.8(139/164) |
| プラセボ群との差[95%信頼区間] | 32.3[16.8,47.7] | 45.8[31.1,60.5] | − | 55.8[43.7,67.9] |
| オッズ比[95%信頼区間]a) | 3.89[2.04,7.43] | 7.26[3.65,14.41] | − | |
| ACR50%改善率 | 34.1(29/85) | 43.4(36/83) | 8.4(7/83) | 55.5(91/164) |
| ACR70%改善率 | 14.1(12/85) | 28.9(24/83) | 0(0/83) | 33.5(55/164) |
投与12週時までの副作用、重篤な副作用は、以下のとおりであった。主な副作用は、100mg群では鼻咽頭炎4.8%(5/104例)、150mg群では鼻咽頭炎7.8%(8/102例)であった。
| 100mg群 | 150mg群 | プラセボ群 | エタネルセプト群 | |
| 副作用 | 31.7(33/104) | 37.3(38/102) | 28.7(29/101) | 37.5(75/200) |
| 重篤な副作用 | 1.9(2/104) | 1.0(1/102) | 3.0(3/101) | 2.0(4/200) |
17.1.2 国内第3相試験(国内、MTX併用、CL-RAJ4試験)
MTXで効果不十分な関節リウマチ患者(目標例数510例(各群170例))を対象としたプラセボ対照無作為化二重盲検並行群間比較試験を実施した。MTX併用下で、本剤100mg、150mg又はプラセボを1日1回朝食後に経口投与した。主要評価項目は投与12週後のACR20%改善率及び投与28週後の手足のX線スコア(van der Heijde Modified Total Sharp Score;mTSS)のベースラインからの変化量のco-primary endpointとされた。本剤100mg及び150mg群の投与12週後のACR20%改善率はプラセボ群に比べ高く、統計学的に有意な差が認められた。また、本剤100mg及び150mg群の投与28週後のmTSSのベースラインからの平均変化量はプラセボ群に比べ小さく、統計学的に有意な差が認められた26)。[7.1、9.3.2、9.3.3参照]
| 100mg群 | 150mg群 | プラセボ群 | |
| ACR20%改善率 | 58.6(102/174) | 64.4(112/174) | 21.8(37/170) |
| プラセボ群との差[95%信頼区間]a) p値b) | 36.9[26.7,47.0] <0.001 | 42.6[32.6,52.6] <0.001 | − |
| ACR50%改善率 | 29.9(52/174) | 46.0(80/174) | 7.6(13/170) |
| ACR70%改善率 | 12.1(21/174) | 23.6(41/174) | 2.4(4/170) |
| 100mg群(164例) | 150mg群(164例) | プラセボ群(153例) | |
| mTSSの変化量 | 1.62±4.23 | 1.03±2.86 | 3.37±5.46 |
| 中央値(第一四分位点,第三四分位点) | 0.00(0.00,1.50) | 0.00(0.00,1.00) | 1.17(0.00,5.50) |
| p値a) | <0.001 | <0.001 | − |
投与12週時までの副作用、重篤な副作用は、以下のとおりであった。主な副作用は、100mg群では鼻咽頭炎6.3%(11/174例)、150mg群では鼻咽頭炎6.9%(12/174例)であった。
| 100mg群 | 150mg群 | プラセボ群 | |
| 副作用 | 32.8(57/174) | 46.0(80/174) | 27.6(47/170) |
| 重篤な副作用 | 1.7(3/174) | 1.7(3/174) | 1.2(2/170) |
17.1.3 継続投与試験(国際共同、CL-RAJ2試験)
後期第2相試験又は第3相試験を完了した患者のうち、移行基準を満たした患者を対象として、本剤の長期の安全性及び有効性を非盲検下で検討した。開始用量として、後期第2相試験からの移行者は本剤50mg、第3相試験からの移行者は本剤100mgを1日1回朝食後に経口投与した。その後、安全性に問題が無く、効果が不十分な患者に対しては本剤150mg1日1回に増量可とした。また、有害事象が発現した患者では、治験責任医師又は治験分担医師の判断で本剤50mg1日1回への減量を可能とした。試験期間中のACR20%改善率の推移は、24週時:82.1%、48週時:85.7%、72週時:85.4%であった27)。副作用、重篤な副作用は69.5%(586/843例)、9.0%(76/843例)であった。主な副作用は、鼻咽頭炎24.4%(206/843例)、帯状疱疹11.5%(97/843例)、血中クレアチンホスホキナーゼ増加5.5%(46/843例)、気管支炎5.3%(45/843例)、インフルエンザ5.2%(44/843例)及び上気道感染5.1%(43/843例)であった(データカットオフ:2018年5月31日)。[7.1、9.3.2、9.3.3参照]
17.3 その他
18. 薬効薬理
18.1 作用機序
JAKファミリーは、免疫・炎症反応及び造血等に関与するサイトカインや成長因子の受容体の細胞内領域に会合しており、受容体下流の細胞内シグナル伝達において重要な役割を担っている。ペフィシチニブは、JAKファミリーを阻害し、炎症性サイトカインのシグナル伝達や細胞増殖を抑制する。
18.2 JAK阻害活性
ペフィシチニブは、in vitroキナーゼアッセイにおいて、JAK1、JAK2、JAK3及びTYK2の活性を阻害し、そのIC50値はそれぞれ、3.92、5.01、0.71及び4.79nmol/Lである29)30)。
18.3 サイトカインシグナル伝達に対する作用
ペフィシチニブは、JAK1及びJAK3が介在するIL-2刺激によるヒト末梢血単核球からのIL-13、GM-CSF、IFN-γ、TNF-αの産生をそれぞれ、IC50値2.43、2.11、0.203及び15.7nmol/Lで抑制する31)。また、本薬は、ヒトCD4+T細胞及びCD8+T細胞において、IL-6のシグナル伝達をそれぞれ、IC50値49.6及び33.5nmol/Lで抑制し、IFN-αのシグナル伝達をそれぞれ、IC50値23.4及び25.4nmol/Lで抑制する32)(in vitro試験)。
18.4 細胞増殖に対する作用
ペフィシチニブは、JAK1及びJAK3が介在するIL-2刺激によるヒト末梢血T細胞の増殖を抑制し、そのIC50値は18.2nmol/Lである33)。一方で、JAK2のみが介在するエリスロポエチン刺激によるヒト赤白血病細胞株の増殖も抑制するが、そのIC50値は248nmol/Lである34)(in vitro試験)。
18.5 関節炎モデルに対する作用
19. 有効成分に関する理化学的知見
19.1. ペフィシチニブ臭化水素酸塩
| 一般的名称 | ペフィシチニブ臭化水素酸塩 |
|---|---|
| 一般的名称(欧名) | Peficitinib Hydrobromide |
| 化学名 | 4-{[(1R,2s,3S,5s,7s)-5-Hydroxyadamantan-2-yl]amino}-1H-pyrrolo[2,3-b]pyridine-5-carboxamide monohydrobromide |
| 分子式 | C18H22N4O2・HBr |
| 分子量 | 407.30 |
| 物理化学的性状 | ペフィシチニブ臭化水素酸塩は白色〜帯黄白色の結晶又は粉末である。水にやや溶けにくく、エタノール(99.5)に溶けにくい。 |
| KEGG DRUG | 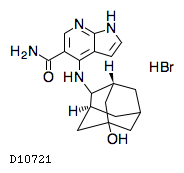 |
20. 取扱い上の注意
本品はアルミ袋、及びアルミ袋に封入している乾燥剤により品質保持をはかっているので、アルミ袋開封後は湿気を避けて保存すること。
21. 承認条件
21.1 医薬品リスク管理計画を策定の上、適切に実施すること。
21.2 製造販売後、一定数の症例に係るデータが集積されるまでの間は、全症例を対象に使用成績調査を実施することにより、本剤の安全性及び有効性に関するデータを早期に収集し、本剤の適正使用に必要な措置を講じること。
22. 包装
<スマイラフ錠50mg>
14錠(7錠×2、乾燥剤入り)
<スマイラフ錠100mg>
14錠(7錠×2、乾燥剤入り)
23. 主要文献
- 社内報告書:ラット、ウサギ・生殖発生毒性試験(2019年3月26日承認 CTD2.6.6.6.1〜2.6.6.6.5)
- 社内報告書:乳汁中への移行・薬物動態試験(2019年3月26日承認 CTD2.6.4.5.6)
- 社内報告書:ラット・生殖発生毒性試験(2019年3月26日承認 CTD2.6.6.6.5)
- 社内報告書:ラット・がん原性試験(2019年3月26日承認 CTD2.6.6.5.6、2.6.6.9.7)
- 社内報告書:健康成人・第1相試験(2019年3月26日承認 CTD2.7.6.9)
- 社内報告書:薬物相互作用試験・メトホルミン(2019年3月26日承認 CTD2.7.6.17、2.7.2.3.1.2)
- 社内報告書:健康成人・食事の影響試験(2019年3月26日承認 CTD2.7.6.1)
- 社内報告書:ヒト血漿蛋白結合率及び結合蛋白の推定・薬物動態試験(2019年3月26日承認 CTD2.7.2.2.1.1、2.7.2.3.3)
- 社内報告書:代謝物の同定及び構造推定・薬物動態試験(2019年3月26日承認 CTD2.7.2.2.1.21、2.7.2.3.4)
- 社内報告書:代謝酵素の同定・薬物動態試験(2019年3月26日承認 CTD2.7.2.2.1.3)
- 社内報告書:健康成人・マスバランス試験(2019年3月26日承認 CTD2.7.6.11)
- 社内報告書:腎機能障害患者試験(2019年3月26日承認 CTD2.7.2.2.2.14)
- 社内報告書:肝機能障害患者試験(2019年3月26日承認 CTD2.7.2.2.2.15)
- 社内報告書:P-gpに対する基質性の検討・薬物動態試験(2019年3月26日承認 CTD2.7.2.2.1.6)
- 社内報告書:薬物相互作用試験・ベラパミル(2019年3月26日承認 CTD2.7.6.14)
- 社内報告書:薬物相互作用試験・メトトレキサート(2019年3月26日承認 CTD2.7.6.18)
- 社内報告書:CYP分子種に対する阻害作用・薬物動態試験(2019年3月26日承認 CTD2.7.2.2.1.4)
- 社内報告書:BCRPに対する阻害作用の検討・薬物動態試験(2019年3月26日承認 CTD2.7.2.2.1.9)
- 社内報告書:OATP1B1及びOATP1B3に対する阻害作用の検討・薬物動態試験(2019年3月26日承認 CTD2.7.2.2.1.15)
- 社内報告書:OCT1及びOCT2に対する阻害作用の検討・薬物動態試験(2019年3月26日承認 CTD2.7.2.2.1.19)
- 社内報告書:薬物相互作用試験・ミダゾラム(2019年3月26日承認 CTD2.7.6.15)
- 社内報告書:薬物相互作用試験・ロスバスタチン(2019年3月26日承認 CTD2.7.6.16)
- 社内報告書:薬物相互作用試験・ミコフェノール酸モフェチル(2019年3月26日承認 CTD2.7.6.19)
- 社内報告書:薬物相互作用試験・タクロリムス(2019年3月26日承認 CTD2.7.6.20)
- 社内報告書:関節リウマチ患者・第3相試験(2019年3月26日承認 CTD2.7.6.24)
- 社内報告書:関節リウマチ患者・第3相試験(2019年3月26日承認 CTD2.7.6.25)
- 社内報告書:関節リウマチ患者・継続投与試験(2019年3月26日承認 CTD2.7.6.28)
- 社内報告書:健康成人・QT/QTc評価試験(2019年3月26日承認 CTD2.7.6.22)
- 社内報告書:薬効薬理試験・JAKキナーゼアッセイ(2019年3月26日承認 CTD2.6.2.2.1)
- 社内報告書:薬効薬理試験・各種キナーゼアッセイ(2019年3月26日承認 CTD2.6.2.2.1)
- 社内報告書:薬効薬理試験・サイトカイン産生(2019年3月26日承認 CTD2.6.2.2.4)
- 社内報告書:薬効薬理試験・サイトカインシグナル伝達(2019年3月26日承認 CTD2.6.2.2.5)
- 社内報告書:薬効薬理試験・T細胞増殖(2019年3月26日承認 CTD2.6.2.2.3)
- 社内報告書:薬効薬理試験・赤白血病細胞増殖(2019年3月26日承認 CTD2.6.2.3.1)
- 社内報告書:薬効薬理試験・ラット関節炎モデル予防的投与(2019年3月26日承認 CTD2.6.2.2.6)
- 社内報告書:薬効薬理試験・ラット関節炎モデル治療的投与(2019年3月26日承認 CTD2.6.2.2.6)
24. 文献請求先及び問い合わせ先
文献請求先
アステラス製薬株式会社 メディカルインフォメーションセンター
〒103-8411 東京都中央区日本橋本町2丁目5番1号
電話:フリーダイヤル 0120-189-371
製品情報問い合わせ先
アステラス製薬株式会社 メディカルインフォメーションセンター
〒103-8411 東京都中央区日本橋本町2丁目5番1号
電話:フリーダイヤル 0120-189-371
26. 製造販売業者等
26.1 製造販売
アステラス製薬株式会社
東京都中央区日本橋本町2丁目5番1号
填写下方邮箱进行订阅,即可获取详细说明书,另有机会获得全处方辅料含量及部分COA等!
更多信息
你必须登录或注册一个新账户,以便接触產品
有用的信息
- 仅对制药厂、医药研发公司、医药研究所、医院服务,不对个人开放!










必须要注册或登陆才可以发表评论
我们提供全球参比制剂,有需要可以评论留言
登录 注册