
 请用手机扫二维码
请用手机扫二维码
医薬品情報
| 総称名 | エベレンゾ |
|---|---|
| 一般名 | ロキサデュスタット |
| 欧文一般名 | Roxadustat |
| 製剤名 | ロキサデュスタット錠 |
| 薬効分類名 | HIF-PH阻害薬 腎性貧血治療薬 |
| 薬効分類番号 | 3999 |
| ATCコード | B03XA05 |
| KEGG DRUG | D10593 ロキサデュスタット |
| JAPIC | 添付文書(PDF) |
添付文書情報2022年11月 改訂(第7版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| エベレンゾ錠20mg | Evrenzo Tablets 20mg | アステラス製薬 | 3999047F1028 | 367.7円/錠 | 劇薬, 処方箋医薬品 |
| エベレンゾ錠50mg | Evrenzo Tablets 50mg | アステラス製薬 | 3999047F2024 | 758.7円/錠 | 劇薬, 処方箋医薬品 |
| エベレンゾ錠100mg | Evrenzo Tablets 100mg | アステラス製薬 | 3999047F3020 | 1370.5円/錠 | 劇薬, 処方箋医薬品 |
1. 警告
本剤投与中に、脳梗塞、心筋梗塞、肺塞栓等の重篤な血栓塞栓症があらわれ、死亡に至るおそれがある。本剤の投与開始前に、脳梗塞、心筋梗塞、肺塞栓等の合併症及び既往歴の有無等を含めた血栓塞栓症のリスクを評価した上で、本剤の投与の可否を慎重に判断すること。また、本剤投与中は、患者の状態を十分に観察し、血栓塞栓症が疑われる徴候や症状の発現に注意すること。血栓塞栓症が疑われる症状があらわれた場合には、速やかに医療機関を受診するよう患者を指導すること。[11.1.1参照]
2. 禁忌
次の患者には投与しないこと
2.1 本剤の成分に対し過敏症の既往歴のある患者
2.2 妊婦又は妊娠している可能性のある女性[9.5参照]
4. 効能または効果
5. 効能または効果に関連する注意
赤血球造血刺激因子製剤で未治療の場合、本剤投与開始の目安は、腹膜透析患者及び保存期慢性腎臓病患者ではヘモグロビン濃度で11g/dL未満、血液透析患者ではヘモグロビン濃度で10g/dL未満とする。
6. 用法及び用量
赤血球造血刺激因子製剤で未治療の場合
通常、成人には、ロキサデュスタットとして1回50mgを開始用量とし、週3回経口投与する。以後は、患者の状態に応じて投与量を適宜増減するが、最高用量は1回3.0mg/kgを超えないこととする。
赤血球造血刺激因子製剤から切り替える場合
通常、成人には、ロキサデュスタットとして1回70mg又は100mgを開始用量とし、週3回経口投与する。以後は、患者の状態に応じて投与量を適宜増減するが、最高用量は1回3.0mg/kgを超えないこととする。
7. 用法及び用量に関連する注意
7.1 赤血球造血刺激因子製剤から切り替える場合の開始用量
下表を参考に切替え前の赤血球造血刺激因子製剤投与量から本剤の投与量を決定し、切り替えること。
| エリスロポエチン製剤(IU/週) | ダルベポエチンアルファ(μg/週) | エポエチンベータペゴル(μg/4週) | 本剤(mg/回) |
| 4500未満 | 20未満 | 100以下 | 70 |
| 4500以上 | 20以上 | 100超 | 100 |
7.2 投与量調整
用量調整が必要な場合には、下表[投与量増減表]、[投与量調整表]を参考に投与量を増減すること。なお、用量調整を行った場合は、少なくとも4週間は同一用量を維持すること。ただし、増量後4週以内にヘモグロビン濃度が急激に上昇(2.0g/dLを超える)した場合、速やかに減量又は休薬すること。[8.1参照]
| 4週前から当該週までのHb値変化量 | 当該週のHb値 | |||
| 10.5g/dL未満 | 10.5g/dL以上 11.5g/dL以下 | 11.5g/dL超 12.5g/dL以下 | 12.5g/dLを超える | |
| −1.0g/dL未満 | 1段階増量 | 1段階増量 | 変更なし | 休薬し、Hb値が11.0g/dL未満になった時点から1段階減量して再開 |
| −1.0g/dL以上 1.0g/dL以下 | 1段階増量 | 変更なし | 1段階減量 | |
| 1.0g/dL超 2.0g/dL以下 | 変更なし | 1段階減量 | 1段階減量 | |
| 2.0g/dLを超える | 1段階減量 | |||
| 段階 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
| 本剤投与量(注) | 20mg | 40mg | 50mg | 70mg | 100mg | 120mg | 150mg | 200mg |
7.3 週3回投与
2〜3日に1回の間隔(例えば月・水・金、又は火・木・土等)で週3回投与すること。
7.4 本剤の服用を忘れた場合
次のあらかじめ定めた日の服用時間帯と24時間以上間隔があく場合は、直ちに服用すること。ただし、以後はあらかじめ定めた日に服用すること。次のあらかじめ定めた日の服用時間帯との間隔が24時間未満である場合は服用せずに、次のあらかじめ定めた日に服用すること。同日に2回分を服用しないこと。
8. 重要な基本的注意
8.1 本剤投与開始後及び用量変更後には、ヘモグロビン濃度が目標範囲に到達し、安定するまでは週1回から2週に1回程度ヘモグロビン濃度を確認すること。ヘモグロビン濃度が4週以内に2.0g/dLを超えるような急激な上昇を認めた場合は、減量・休薬等の適切な処置をとること。[7.2参照]
8.2 本剤投与中はヘモグロビン濃度等を定期的に確認し、腎性貧血の治療に関する最新の情報を参考にして、必要以上の造血作用があらわれないように十分注意すること。赤血球造血刺激因子製剤の臨床試験においてヘモグロビンの目標値を高く設定した場合に、死亡、心血管系障害及び脳卒中の発現頻度が高くなったとの報告がある1)2)3)。
8.3 本剤投与中に中枢性甲状腺機能低下症があらわれることがあり、投与開始後約2週間であらわれたとの報告もある。本剤投与中は定期的に甲状腺機能検査(TSH、遊離T3、遊離T4)を行うなど、患者の状態を十分に観察すること。[11.1.3参照]
8.4 本剤投与により血圧が上昇する場合があるので、血圧の推移に十分注意しながら投与すること。
8.5 造血には鉄が必要なことから、必要に応じて鉄の補充を行うこと。
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
9.1.1 脳梗塞、心筋梗塞、肺塞栓等の患者、又はそれらの既往歴のある患者
本剤投与により血栓塞栓症を増悪あるいは誘発するおそれがある。
9.1.2 高血圧症を合併する患者
血圧上昇があらわれるおそれがある。
9.1.3 悪性腫瘍を合併する患者
本剤の血管新生亢進作用により悪性腫瘍を増悪させる可能性がある。
9.1.4 増殖糖尿病網膜症、黄斑浮腫、滲出性加齢黄斑変性症、網膜静脈閉塞症等を合併する患者
本剤の血管新生亢進作用により網膜出血があらわれる可能性がある。
9.3 肝機能障害患者
9.3.1 中等度以上の肝機能障害(Child-Pugh分類B及びC)のある患者
9.4 生殖能を有する者
妊娠可能な女性には、本剤投与中及び本剤投与終了後一定期間は適切な避妊を行うよう指導すること。[9.5参照]
9.5 妊婦
妊婦又は妊娠している可能性のある女性には投与しないこと。母動物(ラット)への投与で、本剤は胎児に移行し、本剤の最大臨床用量における曝露量の0.4倍の曝露量で出生児の発達遅延、0.8倍の曝露量で出生児生存率の低値等が報告されている5)6)。[2.2、9.4参照]
9.6 授乳婦
9.7 小児等
本剤では小児等を対象とした有効性及び安全性を指標とした臨床試験は実施していない。
10. 相互作用
相互作用序文
薬物代謝酵素用語
CYP2C8
薬物代謝酵素用語
UGT1A9
薬物代謝酵素用語
BCRP
薬物代謝酵素用語
OATP1B1
薬物代謝酵素用語
OAT1
薬物代謝酵素用語
OAT3
10.2 併用注意
リン結合性ポリマー [16.7.2参照] | 本剤と併用した場合、本剤の作用が減弱するおそれがあるため、併用する場合は、前後1時間以上間隔をあけて本剤を服用すること。 | 本剤をセベラマー炭酸塩と同時投与したところ、本剤のAUCinfが低下した10)。 |
[16.7.2参照] | 本剤と併用した場合、本剤の作用が減弱するおそれがあるため、併用する場合は、前後1時間以上間隔をあけて本剤を服用すること。 | 本剤を酢酸カルシウムと同時投与したところ、本剤のAUCinfが低下した10)。 |
等 [16.7.3参照] | HMG-CoA還元酵素阻害剤による筋障害を増強するおそれがあるため、併用する場合は、患者の状態を慎重に観察すること。 | 本剤をシンバスタチン、ロスバスタチン、アトルバスタチンと併用したところ、これらの薬剤のAUCinfが上昇した11)12)。また、本剤投与2時間前、本剤投与の4又は10時間後にシンバスタチンを投与した際も曝露量が上昇した。 本剤のOATP1B1/BCRP阻害作用により、これらの薬剤の血漿中濃度を上昇させる。 |
[16.7.2参照] | 本剤の作用が増強するおそれがあるため、併用する場合は、本剤の減量を考慮するとともに、患者の状態を慎重に観察すること。 | 本剤をプロベネシドと併用したところ、本剤のAUCinfが上昇した13)。 プロベネシドのUGT/OAT阻害作用により、本剤の血漿中濃度を上昇させる。 |
[16.7.2参照] | 本剤の作用が増強するおそれがあるため、併用する場合は、本剤の減量を考慮するとともに、患者の状態を慎重に観察すること。 | 本剤をゲムフィブロジルと併用したところ、本剤のAUCinfが上昇した14)。 ゲムフィブロジルのCYP2C8/OATP1B1阻害作用により、本剤の血漿中濃度を上昇させる可能性がある。 |
11. 副作用
11.1 重大な副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1.1 血栓塞栓症(2.3%)
脳梗塞(0.5%)、急性心筋梗塞(0.1%)、肺塞栓症(0.1%)、シャント閉塞(0.8%)等の血栓塞栓症があらわれることがある。[1.参照]
11.1.2 痙攣発作(頻度不明)
11.1.3 中枢性甲状腺機能低下症(頻度不明)
血中甲状腺刺激ホルモン(TSH)が正常範囲内又は低値を示す中枢性甲状腺機能低下症があらわれることがある。症状や徴候があらわれた場合には、必要に応じて投与の中止、甲状腺ホルモン製剤の投与などの適切な処置を行うこと。[8.3参照]
11.2 その他の副作用
| 1%以上 | 0.5〜1%未満 | 0.5%未満 | 頻度不明 | |
| 心臓障害 | うっ血性心不全、動悸 | |||
| 内分泌障害 | 甲状腺機能低下症 | |||
| 眼障害 | 網膜出血 | |||
| 胃腸障害 | 嘔吐、下痢、便秘 | 悪心、腹部不快感 | 腹痛、消化不良、胃障害 | |
| 一般・全身障害及び投与部位の状態 | 浮腫、末梢性浮腫、倦怠感 | |||
| 感染症及び寄生虫症 | 結膜炎 | |||
| 傷害、中毒及び処置合併症 | シャント狭窄 | |||
| 臨床検査 | リパーゼ増加 | ALT増加、CK増加 | TSH減少、遊離T3減少、遊離T4減少、血中ビリルビン増加 | |
| 代謝及び栄養障害 | 高カリウム血症、高リン酸塩血症、鉄欠乏、食欲減退、低アルブミン血症 | |||
| 神経系障害 | 浮動性めまい | |||
| 精神障害 | 不眠症 | |||
| 生殖系及び乳房障害 | 女性化乳房 | |||
| 呼吸器、胸郭及び縦隔障害 | 咳嗽、間質性肺疾患 | |||
| 皮膚及び皮下組織障害 | そう痒症 | 全身性剥脱性皮膚炎 | ||
| 血管障害 | 高血圧 | |||
| その他 | 医療機器内血栓 |
12. 臨床検査結果に及ぼす影響
13. 過量投与
13.1 症状
本剤を健康成人に5mg/kg(510mg)まで単回投与した際、一過性の心拍数増加が報告されている。本剤の過量投与によりヘモグロビン濃度が必要以上に増加するおそれがある。
13.2 処置
本剤の減量・休薬等の適切な処置を行うこと。本剤は透析で除去されない。
14. 適用上の注意
14.1 薬剤交付時の注意
PTP包装の薬剤はPTPシートから取り出して服用するよう指導すること。PTPシートの誤飲により、硬い鋭角部が食道粘膜へ刺入し、更には穿孔をおこして縦隔洞炎等の重篤な合併症を併発することがある。
16. 薬物動態
16.1 血中濃度
16.1.1 健康成人
(1)単回投与
健康成人男性(30例)に本剤0.3〜4.0mg/kgを空腹時に単回経口投与したとき、血漿中未変化体濃度の推移及び薬物動態パラメータは下記のとおりであった19)。
本剤単回投与時の平均血漿中未変化体濃度の推移
| 投与量 | 例数 | Cmax(μg/mL) | Tmax(h) | AUCinf(μg・h/mL) | t1/2(h) |
| 0.3mg/kg | 6 | 1.9±0.3 | 3.0(2.0-4.0) | 13.8±3.4 | 8.3±0.9 |
| 1.0mg/kg | 6 | 5.4±1.0 | 2.0(2.0-3.0) | 43.6±11.9 | 8.4±2.3 |
| 2.0mg/kg | 6 | 12.9±2.3 | 2.0(1.0-4.0) | 99.7±13.3 | 9.3±2.1 |
| 3.0mg/kg | 6 | 18.9±3.5 | 2.0(2.0-4.0) | 139.3±18.9 | 9.0±1.9 |
| 4.0mg/kg | 6 | 20.8±2.3 | 3.0(1.0-4.0) | 168.8±22.0 | 8.0±0.5 |
(2)反復投与
健康成人男性(21例)に本剤0.3〜3.0mg/kgを空腹時に週3回2週間反復経口投与したとき、血漿中未変化体濃度の推移及び薬物動態パラメータは下記のとおりであった。いずれの投与量でも速やかに定常状態に達し、蓄積性はほとんど認められなかった19)。
本剤反復投与時の平均血漿中未変化体濃度の推移
| 投与量 | 測定日 | 例数 | Cmax(μg/mL) | Tmax(h) | AUC24(μg・h/mL) | t1/2(h) |
| 0.3mg/kg | 1 | 7 | 1.6±0.2 | 2.0(1.0-3.0) | 10.7±1.3 | 9.9±1.2 |
| 12 | 7 | 1.8±0.4 | 2.0(0.5-3.0) | 12.0±1.6 | 10.4±1.8 | |
| 1.0mg/kg | 1 | 7 | 5.4±1.4 | 2.0(1.0-6.0) | 37.4±4.2 | 9.6±1.1 |
| 12 | 7 | 5.2±0.8 | 4.0(2.0-6.0) | 39.7±3.8 | 9.0±1.6 | |
| 3.0mg/kg | 1 | 7 | 18.8±4.2 | 2.0(1.0-2.0) | 138.5±32.9 | 9.9±2.4 |
| 12 | 7 | 18.1±2.5 | 2.0(1.0-4.0) | 153.1±31.9 | 9.7±1.1 |
16.1.2 血液透析患者
血液透析患者(12例)に本剤1.0mg/kg又は2.0mg/kgを空腹時に単回経口投与したとき、透析前(透析2.5時間前)投与と透析後投与で本剤の薬物動態に顕著な差はなく、透析の影響はわずかであった20)。
| 投与量 | 投与タイミング | 例数 | Cmax(μg/mL) | Tmax(h) | AUC24(μg・h/mL) | t1/2(h) |
| 1.0mg/kg | 透析前 | 6 | 4.4±0.5 | 2.5(2.0-4.0) | 40.2±5.5 | 15.8±5.1 |
| 透析後 | 6 | 5.6±1.3 | 1.5(1.0-6.0) | 45.8±5.2 | 15.5±3.1 | |
| 2.0mg/kg | 透析前 | 6 | 9.4±1.2 | 3.0(1.0-4.0) | 90.8±30.4 | 16.2±7.1 |
| 透析後 | 6 | 13.0±2.0 | 2.5(1.0-4.0) | 103.4±27.7 | 20.9±10.5 |
16.1.3 腹膜透析患者
16.1.4 保存期慢性腎臓病患者
母集団薬物動態解析の結果、保存期慢性腎臓病患者と透析患者の薬物動態に明確な差はみられなかった22)。
16.2 吸収
本剤を空腹時に経口投与したとき、おおむね投与2時間後にCmaxに到達する。
健康成人男性(16例)に本剤100mgを食後に単回経口投与したときのCmaxの平均値は、空腹時単回経口投与と比較して20%低値であったが、AUCinfへの影響はわずかであった23)。
健康成人男性(16例)に本剤100mgを食後に単回経口投与したときのCmaxの平均値は、空腹時単回経口投与と比較して20%低値であったが、AUCinfへの影響はわずかであった23)。
16.3 分布
血漿蛋白結合率が非常に高く(約99%)、主にアルブミンと結合した24)(in vitro試験)。
16.4 代謝
健康成人男性(6例)に200mgの[14C]-ロキサデュスタットを空腹時に単回経口投与したとき、ヒト血漿中の主成分は未変化体であった。代謝物は4'-水酸化体の硫酸抱合体、4-O-β-グルクロン酸抱合体、4-O-β-グルコース抱合体が認められ、これら代謝物の曝露量はいずれも薬物由来物質の総曝露量の10%未満であった25)(外国人データ)。
ロキサデュスタットの水酸化代謝は主にCYP2C8が寄与し、4-O-β-グルクロン酸抱合代謝は主にUGT1A9が寄与する7)8)(in vitro試験)。[10.参照]
ロキサデュスタットの水酸化代謝は主にCYP2C8が寄与し、4-O-β-グルクロン酸抱合代謝は主にUGT1A9が寄与する7)8)(in vitro試験)。[10.参照]
16.5 排泄
健康成人男性(16例)に本剤100mgを空腹時に単回経口投与したとき、未変化体の尿中排泄率は約1%であった23)。
健康成人男性(6例)に200mgの[14C]-ロキサデュスタットを空腹時に単回経口投与したとき、投与後192時間までの放射能の尿及び糞中排泄率はそれぞれ約46%及び約50%であった26)(外国人データ)。
健康成人男性(6例)に200mgの[14C]-ロキサデュスタットを空腹時に単回経口投与したとき、投与後192時間までの放射能の尿及び糞中排泄率はそれぞれ約46%及び約50%であった26)(外国人データ)。
16.6 特定の背景を有する患者
16.6.1 腎機能障害患者
本剤100mgを重度の腎機能障害患者(eGFR<30mL/min/1.73m2)9例に単回投与した際のAUCinfの平均値は、腎機能正常被験者と比較して2.2倍上昇した。Cmaxの平均値には明確な差はみられなかった27)(外国人データ)。
16.6.2 肝機能障害患者
本剤100mgを中等度の肝機能障害患者(Child-Pugh分類B)8例に単回投与した際の血漿中非結合型のCmax及びAUCinfの平均値は、肝機能正常被験者と比較してそれぞれ16%高値及び70%高値であった4)(外国人データ)。[9.3.1参照]
16.6.3 高齢者
高齢健康被験者(65歳以上)24例に本剤50〜200mgを単回投与した際のCmax及びAUCinfの平均値は、非高齢健康被験者(18〜45歳)と比較してそれぞれ15%高値及び23%高値であった28)(外国人データ)。
16.7 薬物相互作用
16.7.1 In vitro試験
ロキサデュスタットはCYP2C8、UGT1A9、BCRP、OATP1B1、OAT1及びOAT3の基質であり、CYP2C8(Ki値16.0μM)、BCRP(IC50値3.05μM)及びOATP1B1(IC50値2.59μM)に対して阻害作用を有する7)8)9)29)。[10.参照]
16.7.2 本剤の薬物動態に及ぼす併用薬の影響
(1)リン吸着薬
本剤とセベラマー炭酸塩又は酢酸カルシウムの併用により、本剤のAUCinf及びCmaxは低下した。本剤をリン吸着薬の投与の少なくとも1時間前又は1時間後に投与すると、本剤のCmax及びAUCinfの低下は軽減された10)(外国人データ)。
本剤と炭酸ランタン水和物の併用によって、本剤のAUCinfは12%低下し、Cmaxは影響を受けなかった30)。[10.2参照]
本剤と炭酸ランタン水和物の併用によって、本剤のAUCinfは12%低下し、Cmaxは影響を受けなかった30)。[10.2参照]
| リン吸着薬 | リン吸着薬投与量 | 本剤投与量 | 本剤の投与タイミング | 例数 | 幾何平均比(90%信頼区間) (リン吸着薬併用投与時/ロキサデュスタット単独投与時) | |
| Cmax | AUCinf | |||||
| セベラマー炭酸塩 | 2400mg 1日3回投与 | 200mg 単回投与 | 同時投与 | 24 | 0.34(0.31,0.38) | 0.33(0.31,0.36) |
| リン吸着薬投与1時間前 | 30 | 0.74(0.68,0.82) | 0.59(0.56,0.63) | |||
| リン吸着薬投与1時間後 | 30 | 0.88(0.79,0.97) | 0.76(0.72,0.81) | |||
| 酢酸カルシウム | 1900mg 1日3回投与 | 同時投与 | 24 | 0.48(0.43,0.54) | 0.54(0.49,0.58) | |
| リン吸着薬投与1時間前 | 30 | 0.81(0.73,0.89) | 0.69(0.65,0.73) | |||
| リン吸着薬投与1時間後 | 30 | 0.98(0.89,1.07) | 0.83(0.78,0.88) | |||
(2)その他の薬剤
オメプラゾール(プロトンポンプ・インヒビター、外国人データ)、クレメジン(球形吸着炭)は本剤の薬物動態に対して影響を与えなかった31)32)。
本剤の薬物動態に対するその他の併用薬の影響は下表のとおりであった13)14)(外国人データ)。[10.2参照]
本剤の薬物動態に対するその他の併用薬の影響は下表のとおりであった13)14)(外国人データ)。[10.2参照]
| 併用薬 | 併用薬投与量 | 本剤投与量 | 例数 | 幾何平均比(90%信頼区間) (相互作用薬併用投与時/単独投与時) | |
| Cmax | AUCinf | ||||
| ゲムフィブロジル(国内未承認) (CYP2C8及びOATP1B1阻害剤) | 600mg 1日2回投与 | 100mg 単回投与 | 18 | 1.37(1.29,1.46) | 2.35(2.15,2.56) |
| プロベネシド (UGT、OAT1及びOAT3阻害剤) | 500mg 1日2回投与 | 18 | 1.38(1.22,1.56) | 2.25(2.14,2.37) | |
16.7.3 本剤が併用薬の薬物動態に及ぼす影響
(1)HMG-CoA還元酵素阻害剤(OATP1B1/BCRP基質)
本剤とシンバスタチン、シンバスタチンの活性代謝物(アシド体)、ロスバスタチン又はアトルバスタチンの併用により、これらのAUCinf及びCmaxは上昇した。
シンバスタチンを本剤投与の2時間前、4又は10時間後に投与したところ、同時投与時と同様に、シンバスタチンのAUCinf及びCmaxは上昇した11)12)(外国人データ)。[10.2参照]
シンバスタチンを本剤投与の2時間前、4又は10時間後に投与したところ、同時投与時と同様に、シンバスタチンのAUCinf及びCmaxは上昇した11)12)(外国人データ)。[10.2参照]
| 併用薬 | 併用薬投与量 | 本剤投与量 | HMG-CoA還元酵素阻害剤投与のタイミング | 例数 | 幾何平均比(90%信頼区間) (本剤併用投与時/単独投与時) | |
| Cmax | AUCinf | |||||
| シンバスタチン | シンバスタチンを40mg 単回投与 | 200mg 隔日投与 | 同時投与 | 28 | 1.87(1.56,2.23) | 1.75(1.47,2.09) |
| 本剤投与2時間前 | 24 | 2.32(1.92,2.79) | 1.68(1.44,1.96) | |||
| 本剤投与4時間後 | 24 | 3.10(2.57,3.74) | 1.74(1.50,2.03) | |||
| 本剤投与10時間後 | 24 | 2.39(1.98,2.87) | 1.56(1.34,1.82) | |||
| シンバスタチンアシド体(代謝物) | 同時投与 | 28 | 2.76(2.34,3.24) | 1.85(1.54,2.23) | ||
| 本剤投与2時間前 | 24 | 2.34(1.99,2.76) | 1.89(1.62,2.21) | |||
| 本剤投与4時間後 | 24 | 5.98(5.08,7.04) | 3.42(2.94,3.99) | |||
| 本剤投与10時間後 | 24 | 3.37(2.86,3.97) | 2.51(2.16,2.93) | |||
| ロスバスタチン | 10mg 単回投与 | 同時投与 | 28 | 4.47(3.86,5.18) | 2.93(2.63,3.25) | |
| アトルバスタチン | 40mg 単回投与 | 同時投与 | 24 | 1.34(1.11,1.63) | 1.96(1.71,2.26) | |
(2)その他の薬剤
17. 臨床成績
17.1 有効性及び安全性に関する試験
17.1.1 国内第III相比較試験(血液透析、赤血球造血刺激因子製剤(ESA:erythropoiesis stimulating agents)からの切替え)
血液透析施行中の腎性貧血患者302例(本剤150例、ダルベポエチンアルファ152例)を対象に、前治療のESAの用量に応じて本剤70mg又は100mgから開始し、Hb値に応じて用量を20〜300mgの間で調整し、週3回24週間経口投与した。また、実薬対照としてダルベポエチンアルファを設定した。その結果、投与18週から24週の平均Hb値のベースラインからの変化量の調整済み平均値は、本剤投与群で−0.04g/dL、ダルベポエチンアルファ投与群で−0.03g/dLであり、本剤のダルベポエチンアルファに対する非劣性が検証された15)。
副作用発現頻度は、本剤投与群で22.0%(33/150例)、主な副作用は、高血圧3.3%(5/150例)、嘔吐、網膜出血、低アルブミン血症各2.0%(3/150例)であった。
17.1.2 国内長期投与試験(血液透析、ESAからの切替え)
血液透析施行中の腎性貧血患者163例を対象に、前治療のESAの用量に応じて本剤70mg又は100mgから開始し、Hb値に応じて用量を20〜300mgの間で調整し、週3回52週間経口投与した。その結果、投与46週から52週の目標Hb値維持率(平均Hb値が10.0g/dL以上12.0g/dL以下であった患者割合)は71.2%(116/163例)であった16)。
副作用発現頻度は、27.6%(45/163例)であった。主な副作用は、嘔吐3.1%(5/163例)、腹部不快感、シャント閉塞各2.5%(4/163例)であった。
17.1.3 国内一般臨床試験(血液透析、ESA未治療)
ESA未治療の血液透析施行中の腎性貧血患者75例を対象に、本剤50mg又は70mgから開始し、Hb値に応じて用量を20〜300mgの間で調整し、週3回24週間経口投与した。その結果、投与終了時までの累積奏効率(Hb値が10.0g/dL以上を達成、かつベースラインよりHb値が1.0g/dL以上上昇した患者割合)は、本剤50mg開始群で86.5%(32/37例)、本剤70mg開始群で89.2%(33/37例)であった17)。
副作用発現頻度は、21.3%(16/75例)であった。主な副作用は、シャント閉塞、リパーゼ増加各2.7%(2/75例)であった。
17.1.4 国内一般臨床試験(腹膜透析)
腹膜透析施行中の腎性貧血患者56例(ESA未治療の患者13例、ESAからの切替え患者43例)を対象に、ESA未治療の患者には本剤50mg又は70mgを、ESAからの切替え患者には前治療のESAの用量に応じて本剤70mg又は100mgから開始し、Hb値に応じて用量を20〜300mgの間で調整し、週3回24週間経口投与した。その結果、投与18週から24週の目標Hb値維持率(平均Hb値が10.0g/dL以上12.0g/dL以下であった患者割合)は、ESA未治療の本剤50mg開始群で83.3%(5/6例)、ESA未治療の本剤70mg開始群で100.0%(7/7例)、ESAからの切替え患者で74.4%(32/43例)であった18)。
副作用発現頻度は、37.5%(21/56例)であった。主な副作用は、便秘、そう痒症各5.4%(3/56例)、下痢、浮腫、結膜炎、ALT増加、咳嗽各3.6%(2/56例)であった。
17.1.5 国内第III相比較試験(保存期、ESAからの切替え)
保存期慢性腎臓病の腎性貧血患者332例(遺伝子組換えヒトエリスロポエチン(rHuEPO)製剤もしくはダルベポエチンアルファからの切替え群として、本剤投与群131例、ダルベポエチンアルファ投与群131例、また、エポエチンベータペゴルから本剤への切替え群として70例(参照群))を対象に、前治療のESAの用量に応じて本剤70mg又は100mgから開始し、Hb値に応じて用量を20〜300mgの間で調整し、週3回52週間経口投与した。また、実薬対照としてダルベポエチンアルファを設定した。その結果、投与18週から24週の平均Hb値のベースラインからの変化量の調整済み平均値は、本剤投与群で0.15g/dL、ダルベポエチンアルファ投与群で0.22g/dLであり、本剤のダルベポエチンアルファに対する非劣性が検証された36)。
副作用発現頻度は、本剤投与群で17.4%(35/201例)、主な副作用は、高血圧3.0%(6/201例)であった。
17.1.6 国内一般臨床試験(保存期、ESA未治療)
ESA未治療の保存期慢性腎臓病の腎性貧血患者99例を対象に、本剤50mg又は70mgから開始し、Hb値に応じて用量を20〜300mgの間で調整し、週3回24週間経口投与した。その結果、投与終了時までの累積奏効率(Hb値が10.0g/dL以上を達成、かつベースラインよりHb値が1.0g/dL以上上昇した患者割合)は、本剤50mg開始群で93.9%(46/49例)、本剤70mg開始群で100.0%(50/50例)であった37)。
副作用発現頻度は、14.1%(14/99例)であった。主な副作用は、高血圧2.0%(2/99例)であった。
18. 薬効薬理
18.1 作用機序
ロキサデュスタットは、転写因子である低酸素誘導因子(HIF:hypoxia inducible factor)の分解に関わるHIF-プロリン水酸化酵素(HIF-PH)を阻害する38)。それにより、HIF-αの分解が妨げられてHIF経路が活性化され、その結果、エリスロポエチンが増加することにより、赤血球形成が促進されると考えられる。
18.2 HIF-PH阻害作用及びエリスロポエチン産生作用
ロキサデュスタットは、in vitroにおいてHIF-PHであるプロリン水酸化酵素ドメイン(PHD)1、PHD2、及びPHD3を阻害した38)。また、ヒト肝細胞培養系においてHIF-2α蛋白の蓄積を誘導し、エリスロポエチン産生を増加させた39)。
18.3 貧血改善作用
19. 有効成分に関する理化学的知見
19.1. ロキサデュスタット
| 一般的名称 | ロキサデュスタット |
|---|---|
| 一般的名称(欧名) | Roxadustat |
| 化学名 | N-[(4-Hydroxy-1-methyl-7-phenoxyisoquinolin-3-yl)carbonyl]glycine |
| 分子式 | C19H16N2O5 |
| 分子量 | 352.34 |
| 物理化学的性状 | ロキサデュスタットは白色〜黄色の結晶又は粉末である。水にほとんど溶けず、エタノール(99.5)に溶けにくい。 |
| KEGG DRUG | 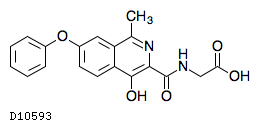 |
21. 承認条件
医薬品リスク管理計画を策定の上、適切に実施すること。
22. 包装
<エベレンゾ錠20mg>
30錠(3錠×10)
<エベレンゾ錠50mg>
30錠(3錠×10)
<エベレンゾ錠100mg>
30錠(3錠×10)
23. 主要文献
- Besarab,A.et al., N.Engl.J.Med., 339, 584-590, (1998) »PubMed »J-STAGE
- Singh,A.K.et al., N.Engl.J.Med., 355, 2085-2098, (2006) »PubMed »J-STAGE
- Pfeffer,M.A.et al., N.Engl.J.Med., 361, 2019-2032, (2009) »PubMed »J-STAGE
- 社内報告書:海外肝機能障害患者・薬物動態試験(2019年9月20日承認 CTD 2.7.6.13)
- 社内報告書:ラット・生殖発生毒性試験(2019年9月20日承認 CTD 2.6.6.6.3.1)
- 社内報告書:ラット・乳母哺育試験(2019年9月20日承認 CTD 2.6.6.6.3.2)
- 社内報告書:CYP同定・薬物動態試験(2019年9月20日承認 CTD 2.7.2.2.1.2.7)
- 社内報告書:UGT同定・薬物動態試験(2019年9月20日承認 CTD 2.7.2.2.1.2.8)
- 社内報告書:トランスポーター基質性及び阻害・薬物動態試験(2019年9月20日承認 CTD 2.7.2.2.1.4.3)
- 社内報告書:海外健康成人・薬物相互作用試験(セベラマー及び酢酸カルシウム)(2019年9月20日承認 CTD 2.7.6.18)
- 社内報告書:海外健康成人・薬物相互作用試験(シンバスタチン及びロスバスタチン)(2019年9月20日承認 CTD 2.7.6.22)
- 社内報告書:海外健康成人・薬物相互作用試験(アトルバスタチン)(2019年9月20日承認 CTD 2.7.6.23)
- 社内報告書:海外健康成人・薬物相互作用試験(プロベネシド)(2019年9月20日承認 CTD 2.7.6.21)
- 社内報告書:海外健康成人・薬物相互作用試験(ゲムフィブロジル)(2019年9月20日承認 CTD 2.7.6.16)
- 社内報告書:血液透析患者・二重盲検比較試験(2019年9月20日承認 CTD 2.7.6.28)
- 社内報告書:血液透析患者・長期投与試験(2019年9月20日承認 CTD 2.7.6.34)
- 社内報告書:血液透析患者・ESA未治療患者対象試験(2019年9月20日承認 CTD 2.7.6.33)
- 社内報告書:腹膜透析患者・第III相試験(2019年9月20日承認 CTD 2.7.6.32)
- 社内報告書:健康成人・薬物動態試験(2019年9月20日承認 CTD 2.7.6.4)
- 社内報告書:血液透析患者・薬物動態試験(2019年9月20日承認 CTD 2.7.6.9)
- 社内報告書:透析患者・母集団薬物動態解析(2019年9月20日承認 CTD 2.7.2.2.7.2)
- 社内報告書:腎性貧血患者・母集団薬物動態解析
- 社内報告書:健康成人・食事の影響試験(2019年9月20日承認 CTD 2.7.6.1)
- 社内報告書:血漿蛋白結合・薬物動態試験(2019年9月20日承認 CTD 2.7.2.2.1.1.1)
- 社内報告書:ヒト代謝物プロファイリング・薬物動態試験(2019年9月20日承認 CTD 2.7.2.2.1.2.5)
- 社内報告書:海外健康成人・マスバランス試験(2019年9月20日承認 CTD 2.7.6.8)
- 社内報告書:海外腎機能障害患者・薬物動態試験(2019年9月20日承認 CTD 2.7.6.12)
- 社内報告書:海外健康成人・加齢及び性差検討試験(2019年9月20日承認 CTD 2.7.6.11)
- 社内報告書:CYP阻害・薬物動態試験(2019年9月20日承認 CTD 2.7.2.2.1.3.1)
- 社内報告書:健康成人・薬物相互作用試験(炭酸ランタン)(2019年9月20日承認 CTD 2.7.6.15)
- 社内報告書:海外健康成人・薬物相互作用試験(オメプラゾール)(2019年9月20日承認 CTD 2.7.6.19)
- 社内報告書:健康成人・薬物相互作用試験(クレメジン)(2019年9月20日承認 CTD 2.7.6.14)
- 社内報告書:海外健康成人・薬物相互作用試験(ブプロピオン)(2019年9月20日承認 CTD 2.7.6.20)
- 社内報告書:海外健康成人・薬物相互作用試験(ワルファリン)(2019年9月20日承認 CTD 2.7.6.17)
- 社内報告書:海外健康成人・薬物相互作用試験(ロシグリタゾン)(2019年9月20日承認 CTD 2.7.6.25)
- 社内報告書:保存期慢性腎臓病患者・第III相比較試験
- 社内報告書:保存期慢性腎臓病患者・ESA未治療患者対象試験
- 社内報告書:ヒトリコンビナント酵素・薬理作用(2019年9月20日承認 CTD 2.6.2.2.1)
- 社内報告書:ヒト肝細胞・薬理作用(2019年9月20日承認 CTD 2.6.2.2.2)
- 社内報告書:炎症性貧血モデルラット2週間投与・薬理作用(2019年9月20日承認 CTD 2.6.2.2.6.1)
- 社内報告書:炎症性貧血モデルラット4週間投与・薬理作用(2019年9月20日承認 CTD 2.6.2.2.6.2)
- 社内報告書:腎性貧血モデルラット・薬理作用(2019年9月20日承認 CTD 2.6.2.2.7)
24. 文献請求先及び問い合わせ先
文献請求先
アステラス製薬株式会社 メディカルインフォメーションセンター
〒103-8411 東京都中央区日本橋本町2丁目5番1号
電話:フリーダイヤル 0120-189-371
製品情報問い合わせ先
アステラス製薬株式会社 メディカルインフォメーションセンター
〒103-8411 東京都中央区日本橋本町2丁目5番1号
電話:フリーダイヤル 0120-189-371
26. 製造販売業者等
26.1 製造販売
アステラス製薬株式会社
東京都中央区日本橋本町2丁目5番1号
26.2 提携
FibroGen Inc.
填写下方邮箱进行订阅,即可获取详细说明书,另有机会获得全处方辅料含量及部分COA等!
更多信息
你必须登录或注册一个新账户,以便接触產品
有用的信息
- 仅对制药厂、医药研发公司、医药研究所、医院服务,不对个人开放!










必须要注册或登陆才可以发表评论
我们提供全球参比制剂,有需要可以评论留言
登录 注册